
Forest Tree Adaptation to Climate in Red Spruce and Poplar • Invasive Species • Conservation Genetics
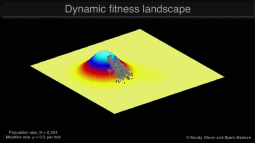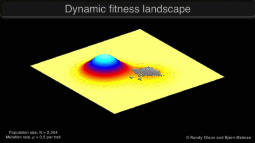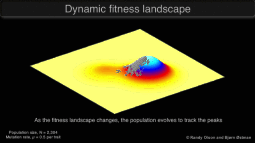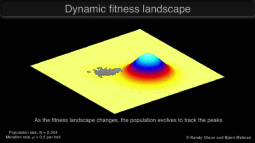
Research in the Keller Lab addresses how populations will be impacted by a rapid change in the selective environment. We approach this question using a variety of study systems experiencing global change.
Graphic © Randy Olson and Bjørn Østman
Research in the Keller Lab addresses how populations will be impacted by a rapid change in the selective environment. We approach this question using a variety of study systems experiencing global change.
Graphic © Randy Olson and Bjørn Østman
Forest Tree Adaptation to Climate
After the end of the Pleistocene ice age, about 20,000 years ago, changing climate caused many species to expand their ranges out of refugial areas to recolonize the vast continental land masses left vacant by retreating glaciers. Major evolutionary and demographic events, such as colonization, population growth, and local adaptation in response to climate changes, are recorded in the genomes of species. We can learn a lot about how species responded to historical climate change by studying the impact of post-glacial warming on genomic diversity and physiological adaptation across the landscape. In addition, knowing the genomic regions associated with adaptation to local climate conditions, especially in ecologically and economically important species such as forest trees, is key to predicting and managing forest responses to current and future climate change.
 collecting spruce cones; see more pictures of spruce research under the Photos tab
collecting spruce cones; see more pictures of spruce research under the Photos tab
Red Spruce
Red spruce (Picea rubens) plays a prominent role in boreal communities throughout the Appalachians, and its populations are marked by distinct evolutionary histories. In a three-year project funded by the National Science Foundation, our lab is studying local adaptation to climate across three zones of P. rubens distribution: the current core of the range in the Northeast, the marginal zone at lower latitudes with more fragmented populations, and the trailing edge of the range, from Pennsylvania to Tennessee, where spruce are isolated on mountaintops. These “island” populations are remnants of spruce forests that covered the South when the ice sheet extended as far south as Long Island. As the glacier retreated and temperatures warmed, spruce retreated to these mountaintop refugia, bringing the rich genetic legacy of these ancient forests with them. The more northern range core, by contrast, was recolonized after the glacier and thus, despite being much larger than the edge populations, has less genetic diversity to draw on and has had less time to adapt.
The goal of our study is to understand the influence of population size, range limit, and genetic diversity on P. rubens local adaptation to climate change in the past, present, and future. With our partners, the University of Maryland Center for Environmental Science and the U.S. Forest Service Southern Research Station in North Carolina, we are planting three common gardens of red spruce collected from 340 mother trees at 65 locations spread throughout the range. We’ll observe how the seedlings respond to the climates of these different latitudes, then link their growth strategies to their population of origin through genetic analysis. We're also sequencing ancient DNA (aDNA) from fossilized red spruce pollen grains that physically span the length of the range and temporally extend back 15,000 years to capture the northern expansion of red spruce during the glacial retreat of the Holocene. We target four loci with intraspecific variation that will give us datasets directly from the past to help us understand how shifting range limits impacted population dynamics during a period of climate warming. Our multifaceted approach will not only paint a clearer scientific picture of genetic adaptation within local populations; it will also inform on-the-ground efforts to restore red spruce to forests where they’ve been lost.
Red spruce (Picea rubens) plays a prominent role in boreal communities throughout the Appalachians, and its populations are marked by distinct evolutionary histories. In a three-year project funded by the National Science Foundation, our lab is studying local adaptation to climate across three zones of P. rubens distribution: the current core of the range in the Northeast, the marginal zone at lower latitudes with more fragmented populations, and the trailing edge of the range, from Pennsylvania to Tennessee, where spruce are isolated on mountaintops. These “island” populations are remnants of spruce forests that covered the South when the ice sheet extended as far south as Long Island. As the glacier retreated and temperatures warmed, spruce retreated to these mountaintop refugia, bringing the rich genetic legacy of these ancient forests with them. The more northern range core, by contrast, was recolonized after the glacier and thus, despite being much larger than the edge populations, has less genetic diversity to draw on and has had less time to adapt.
The goal of our study is to understand the influence of population size, range limit, and genetic diversity on P. rubens local adaptation to climate change in the past, present, and future. With our partners, the University of Maryland Center for Environmental Science and the U.S. Forest Service Southern Research Station in North Carolina, we are planting three common gardens of red spruce collected from 340 mother trees at 65 locations spread throughout the range. We’ll observe how the seedlings respond to the climates of these different latitudes, then link their growth strategies to their population of origin through genetic analysis. We're also sequencing ancient DNA (aDNA) from fossilized red spruce pollen grains that physically span the length of the range and temporally extend back 15,000 years to capture the northern expansion of red spruce during the glacial retreat of the Holocene. We target four loci with intraspecific variation that will give us datasets directly from the past to help us understand how shifting range limits impacted population dynamics during a period of climate warming. Our multifaceted approach will not only paint a clearer scientific picture of genetic adaptation within local populations; it will also inform on-the-ground efforts to restore red spruce to forests where they’ve been lost.
 collecting poplar data in Saskatchewan
collecting poplar data in Saskatchewan
Poplar
Our research integrates tools from genomics, remote sensing, and geospatial modeling to study the genetic basis of climate adaptation in balsam poplar (Populus balsamifera), a key-stone forest tree species in North America with a transcontinental distribution. We have focused on balsam poplar’s southern range edge in order to study the genomics and physiological adaptations of populations with the warmest, earliest onset growing seasons in its geographic range. We've generated genome-wide single nucleotide polymorphism (SNP) data using Genotype by Sequencing (GBS) and targeted capture of flowering-time genes on hundreds of poplar genotypes sampled across the range, and we have used these to perform genome scans for local adaptation. We are also pairing the genomic data with phenotypes measured in common gardens here in Vermont and in Saskatchewan to investigate quantitative genetic evidence of local adaptation to climate and to perform genome-wide association studies (GWAS) for phenology, growth, and water-use efficiency traits.
One of the interesting outcomes of these analyses is that we have detected extensive introgression in range-edge populations of Populus where congeners overlap. These hybrid zones appear to contribute significantly to the adaptive potential of range-edge populations. Regions of the genome associated with climate adaptation can predict field performance via an independent sample of genotypes and an innovative remote sensing approach to measuring phenology. We are also developing new spatial analytical methods to characterize the associations between genomic variation and environmental gradients of climate and growing season length, and to visualize the landscape surface of adaptive variation under both current and projected climates.
Our research integrates tools from genomics, remote sensing, and geospatial modeling to study the genetic basis of climate adaptation in balsam poplar (Populus balsamifera), a key-stone forest tree species in North America with a transcontinental distribution. We have focused on balsam poplar’s southern range edge in order to study the genomics and physiological adaptations of populations with the warmest, earliest onset growing seasons in its geographic range. We've generated genome-wide single nucleotide polymorphism (SNP) data using Genotype by Sequencing (GBS) and targeted capture of flowering-time genes on hundreds of poplar genotypes sampled across the range, and we have used these to perform genome scans for local adaptation. We are also pairing the genomic data with phenotypes measured in common gardens here in Vermont and in Saskatchewan to investigate quantitative genetic evidence of local adaptation to climate and to perform genome-wide association studies (GWAS) for phenology, growth, and water-use efficiency traits.
One of the interesting outcomes of these analyses is that we have detected extensive introgression in range-edge populations of Populus where congeners overlap. These hybrid zones appear to contribute significantly to the adaptive potential of range-edge populations. Regions of the genome associated with climate adaptation can predict field performance via an independent sample of genotypes and an innovative remote sensing approach to measuring phenology. We are also developing new spatial analytical methods to characterize the associations between genomic variation and environmental gradients of climate and growing season length, and to visualize the landscape surface of adaptive variation under both current and projected climates.
Invasive Species
 collecting data on Centaurea in the greenhouse
collecting data on Centaurea in the greenhouse
Invasion of numerous exotic plant species has been preceded or accompanied by interspecific hybridization, suggesting that hybridization could be a key evolutionary mechanism that stimulates invasiveness. Admixture between parental genomes may increase the fitness of hybrids through short-term hybrid vigor and/or increased evolutionary potential in later recombinant generations. However, many plant taxa capable of hybridizing also show partial reproductive isolation because of genetic incompatibilities. Consequently, the ways in which hybridization may facilitate invasion depend on whether hybrid genotypes are restricted to early generations (or maintained through clonal propagation), or rather if hybrid genomes are porous to introgression through advanced generations of recombination. Despite widespread appreciation of the association between hybridization and invasion, few studies have actually tested whether this association reflects short-term versus advanced generations of introgression.
Knapweed (Centaurea spp.), an invasive weed native to Europe, abounds along roadsides and in agricultural fields throughout the Northeast. Our work has shown that two different knapweed species, C. nigra and C. jacea, hybridize extensively in North America as C. x moncktonii. Our research aims to quantify the genomic extent of this introgression and to determine how it affects plant fitness and ultimately whether hybridization is a key component of Centaurea's invasive ability. We have taken several different approaches to these questions. First, we've completed a population genomic analysis using genome-wide SNP data to identify hybrids and examine the extent of genomic introgression between the parent species. In the greenhouse, we have planted common garden experiments to assess the functional traits of plants that vary in their hybrid ancestry, which we are validating using manipulative crosses between C. nigra and C. jacea to generate experimental hybrids. Lastly, we are conducting field studies of population demography to assess what life history stages seem particularly important to population growth rates and whether the traits that are affected by hybridization contribute to increasing population growth rates. In future studies, we aim to use genetic mapping to identify QTL underlying traits linked to invasiveness, and to determine how these evolve under short- and longer-term hybridization. We also plan to broaden our phylogeographic sampling to include more invasive populations as well as the native European range.
In past and ongoing projects on the genetics of invasive species, we have also used campion (Silene spp.) to investigate how founder effects, multiple introductions and admixture, and rapid evolution shape the fate of introduced populations that may become invasive. We have studied reed canary grass (Phalaris arundinacea), in colloboration with Jane Molofsky, to determine how eco-evolutionary feedbacks result in novel genotypes capable of dominating North American wetland ecosystems. With Bob Hilderbrand, we have used environmental DNA (eDNA) as a genetic monitoring system to detect microscopic occurrences of the invasive aquatic diatom Didymosphenia geminata, also known as “rock snot,” in the Cheseapeake Bay watershed.
Knapweed (Centaurea spp.), an invasive weed native to Europe, abounds along roadsides and in agricultural fields throughout the Northeast. Our work has shown that two different knapweed species, C. nigra and C. jacea, hybridize extensively in North America as C. x moncktonii. Our research aims to quantify the genomic extent of this introgression and to determine how it affects plant fitness and ultimately whether hybridization is a key component of Centaurea's invasive ability. We have taken several different approaches to these questions. First, we've completed a population genomic analysis using genome-wide SNP data to identify hybrids and examine the extent of genomic introgression between the parent species. In the greenhouse, we have planted common garden experiments to assess the functional traits of plants that vary in their hybrid ancestry, which we are validating using manipulative crosses between C. nigra and C. jacea to generate experimental hybrids. Lastly, we are conducting field studies of population demography to assess what life history stages seem particularly important to population growth rates and whether the traits that are affected by hybridization contribute to increasing population growth rates. In future studies, we aim to use genetic mapping to identify QTL underlying traits linked to invasiveness, and to determine how these evolve under short- and longer-term hybridization. We also plan to broaden our phylogeographic sampling to include more invasive populations as well as the native European range.
In past and ongoing projects on the genetics of invasive species, we have also used campion (Silene spp.) to investigate how founder effects, multiple introductions and admixture, and rapid evolution shape the fate of introduced populations that may become invasive. We have studied reed canary grass (Phalaris arundinacea), in colloboration with Jane Molofsky, to determine how eco-evolutionary feedbacks result in novel genotypes capable of dominating North American wetland ecosystems. With Bob Hilderbrand, we have used environmental DNA (eDNA) as a genetic monitoring system to detect microscopic occurrences of the invasive aquatic diatom Didymosphenia geminata, also known as “rock snot,” in the Cheseapeake Bay watershed.
Conservation Genetics
|
In addition to the conservation benefits of our research described above, we have collaborated with other scientists to contribute to conservation genetics in the zoological realm. With Dave Nelson, Ed Gates, and Matt Fitzpatrick at the University of Maryland Center for Environmental Science, we have assessed the impact of wind power plants on bats in the central Appalachians using a novel combination of stable isotope and genetic data. We have also studied mating system evolution in social rodents, notably prairie dogs, and fine-scale genetic structure and local adaptation in brook trout.
|

Eastern red bat (Lasiurus borealis)
|

|
